Updated August 19, 2024
Examples of SCGC service deliverables, benchmark data and public data releases can be found here. To complete an SCGC service inquiry or request, start here. For a PDF version of this page, click here.
Table 1. List of SCGC services. See detailed description below.
| Service | Cat # | Unit | Unit price* |
| SAG Generation | |||
| SAG Generation 2 | S-201 | 384-well plate | $3,800 |
| SAG Generation 3 | S-202 | 384-well plate | $7,400 |
| Integrated single cell respiration measurement | S-203 | 384-well plate | $900 |
| Biosafety level 2 handling | S-204 | 384-well plate | $600 |
| SAG SEQUENCING AND BIOINFORMATICS | |||
| SAG WGS 1.8 billion reads | S-211 | ≤384 SAGs | $24,000 |
| SAG WGS 0.4 billion reads | S-212 | ≤384 SAGs | $19,000 |
| SAG WGS 0.1 billion reads | S-216 | ≤384 SAGs | $15,000 |
| SAG WGS addon 1.8 billion reads | S-213 | ≤384 SAGs | $14,000 |
| SAG WGS addon 0.4 billion reads | S-214 | ≤384 SAGs | $9,000 |
| SAG WGS addon 0.1 billion reads | S-215 | ≤384 SAGs | $6,000 |
| Semi-Permeable Capsule (SPC) Library Processing NEW! | |||
| SPC library de novo assembly and annotation | S-301 | 109 PE reads | $3,000 |
| SPC sequencing, assembly and annotation, 0.1 billion reads | S-302 | 1 library | $6,000 |
| SPC sequencing, assembly and annotation, 0.4 billion reads | S-303 | 1 library | $9,000 |
| SPC sequencing, assembly and annotation, 1.8 billion reads | S-304 | 1 library | $14,000 |
| ADDITIONAL SERVICES | |||
| SAG re-arraying | S-105 | 96-well plate | $800 |
| Sample cryoprotectant glyTE | S-019 | 10 mL | $100 |
| Consultation | S-011 | 1 hour | $260 |
| Customized Services | S-100 | Custom | Request a quote |
OVERVIEW
Single cell DNA sequencing
Single cell genomics unveils the genomic blueprints of the most fundamental units of life. It is a powerful approach to analyze biochemical properties, evolutionary histories and the biotechnological potential of uncultured microorganisms, which constitute over 99% of biological diversity on our planet. Single cell genomics is also emerging as a revolutionary technology in the studies of cancer, autoimmune diseases and hereditary disorders. Single cell genomics consists of a series of integrated processes, starting with appropriate sample collection and preservation, followed by physical separation, lysis and whole genome amplification of individual cells, then proceeding to DNA sequencing and sequence interpretation (1). These processes are incorporated in the comprehensive suite of services offered by SCGC (Table 1).
The Single Cell Genomic Center (SCGC®) at Bigelow Laboratory for Ocean Sciences
The SCGC is world’s first research and service center with the primary focus on the single cell genomics of microorganisms (see About SCGC). This includes microorganisms from diverse microbiomes, biosafety level 2 organisms and organisms from hard-to-process environments, such as soil and the deep biosphere. SCGC also has experience and capabilities to process individual cells of humans and other multicellular organisms.
Commitment to Quality
The processing of exceedingly small DNA quantities makes single cell genomics highly susceptible to DNA contamination and amplification biases. At SCGC, we take these risks very seriously and have developed techniques to minimize and monitor methodological artifacts at each point in our workflow. Single cell/particle sorting and DNA amplification are performed in a cleanroom environment, and all associated consumables are decontaminated using methods that were developed and evaluated at SCGC (2). Single amplified genome (SAG) generation services include multiple controls to detect potential DNA contamination. To prevent index switching during SAG sequencing, multiplexed libraries contain dual, 10 bp barcodes. Prior to de novo assembly, raw reads are quality-filtered and digitally normalized using in-house, optimized protocols (2). Genome de novo assemblies are analyzed using multiple QC algorithms. The entire workflow is evaluated for contamination and assembly errors using microbial benchmark cultures with diverse genome complexity and GC content (%), indicating no non-target and undefined bases and average frequencies of mis-assemblies, indels and mismatches at <5 per 100 kbp (2).
SCGC SERVICES: SAG GENERATION
Single amplified genomes (SAGs) are products of whole genome amplification reactions performed on individual cells or other DNA-containing particles (1, 3). SAG generation involves measurement of optical properties, physical separation, lysis and DNA amplification of individual cells/particles. SCGC has developed state-of-the-art techniques for each of these steps (2) and combined them in services S-201 – S-204.
SAG-Generation (S-201)
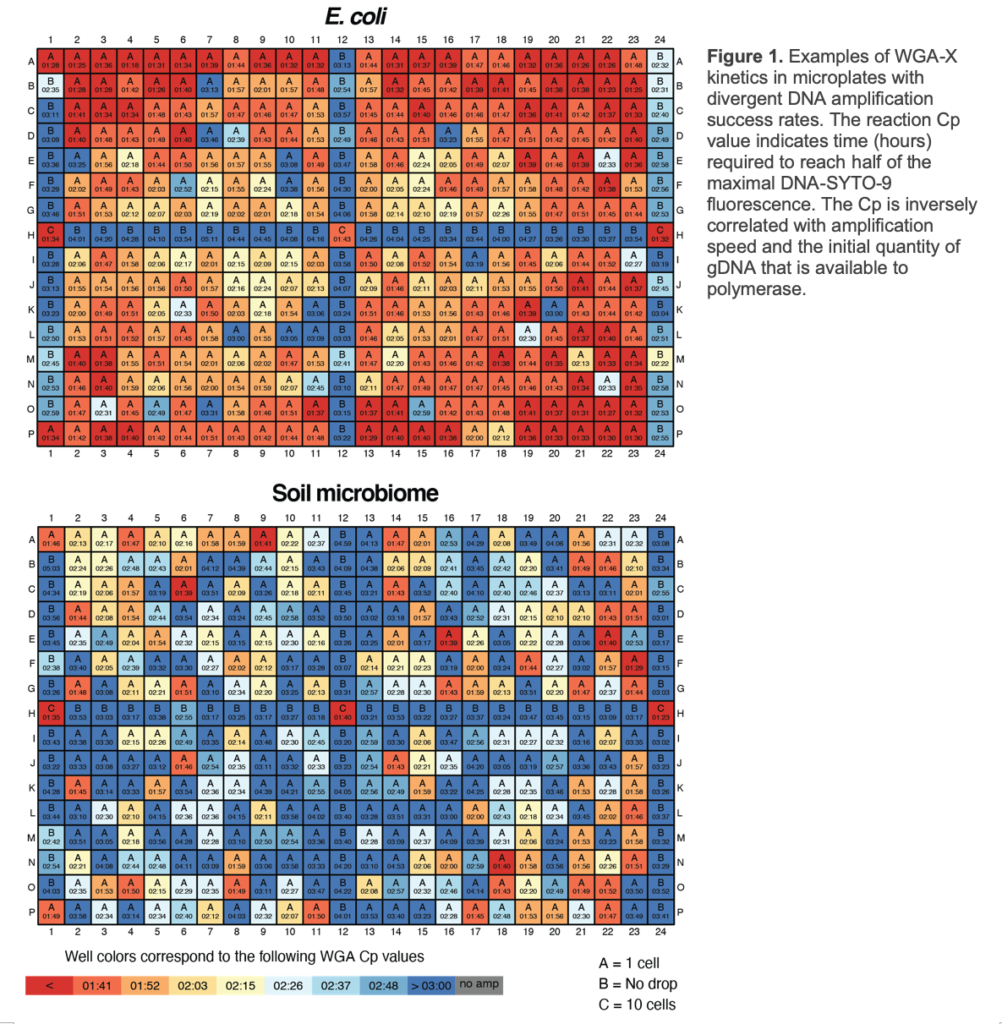
The S-201 service includes separation of individual cells or DNA-containing particles into wells of a 384-well plate by fluorescence-activated cell sorting (FACS), followed by cell lysis and genomic DNA amplification. Cells/particles are separated using an inFlux Mariner (BD), which can be finely tuned to select individual cells or particles based on a range of optical characteristics. Cells/particles may be selected based on the particle autofluorescence, the fluorescent DNA stain SYTO-9 (Thermo Fisher Scientific, provided by SCGC) or other stains and probes that are applied by an SCGC customer prior to shipping to SCGC. During sorting, multiple wells are used as negative and positive controls on each plate (Figure 1).
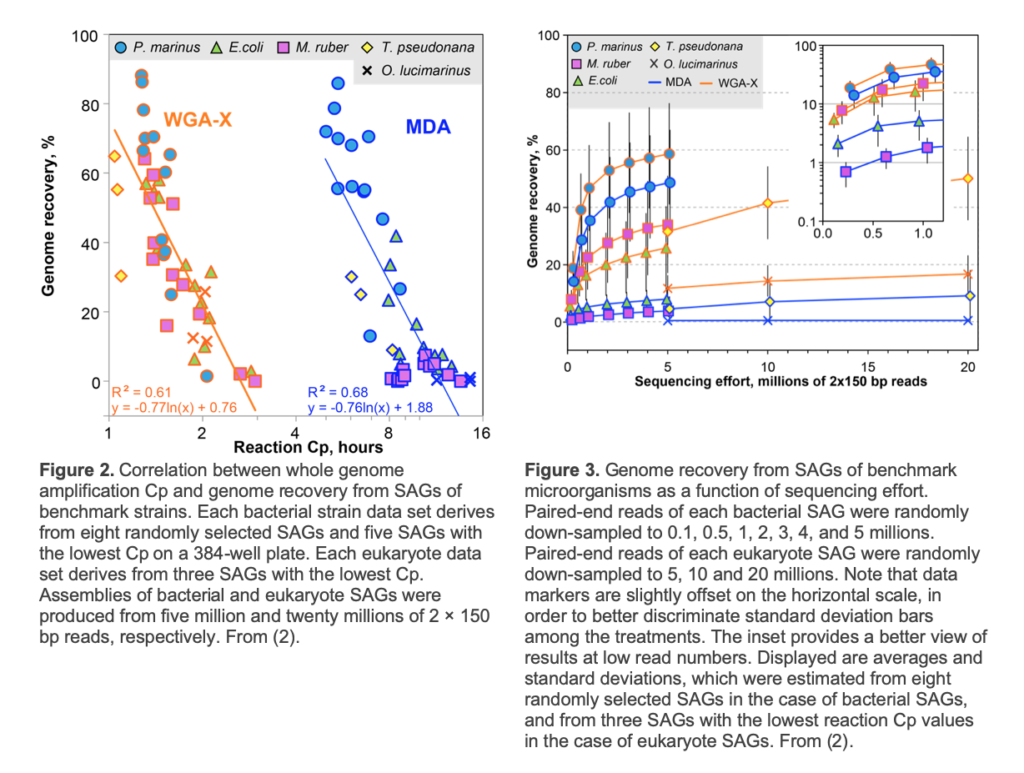
Cells/particles are lysed and their DNA is denatured by 2 freeze-thaw cycles and a subsequent KOH treatment (2). The S-201 service uses WGA-X® for genomic DNA amplification, a method developed by SCGC (2). Compared to the earlier versions of multiple displacement amplification technique (MDA) (4), WGA-X improves average genome recovery from individual cells and viral particles, with most notable enhancements observed in SAGs with high G+C content (Figures 2 and 3). Please note that SAG generation success varies among samples (Figure 1) and depends on many factors, such as: a) prompt cryopreservation of intact cells and gDNA prior to cell sorting; b) successful discrimination of cells and viral particles from other particles during FACS; c) successful single cell and viral particle lysis and DNA amplification. Deliverables of S-201 include:
Deliverables of S-201 include:
a) One 384-well microplate containing WGA-X products of individual cells or DNA-containing particles, 10 uL per well, usually averaging ~1 microgram gDNA per well.
b) Index FACS data files.
c) WGA-X kinetics data obtained by measuring fluorescence of a DNA stain in each well.
For a standard S-201 service fee to be applicable, the following conditions must be met:
a) Target cells or particles are in aquatic solution, are less than 40 micrometers in diameter, and are cryopreserved and shipped following SCGC recommendations (see Preparation and Shipment on SCGC website). Samples that have not been brought into aquatic suspension by SCGC customers may be analyzed as a customized service. Please contact SCGC manager Brian Thompson for additional advice on how to produce SCGC-compatible samples in your own lab or for a feasibility assessment and a quote.
b) Sorting is based on particle autofluorescence, SYTO-9 fluorescence (DNA stain provided by SCGC), or probes that are applied by SCGC customer prior to shipping to SCGC.
c) Only one sample is used per microplate, following SCGC’s standard plate setup (Figure 1).
d) If a sample is known to contain Biosafety Level 2 microorganisms or genetically modified organisms, then an add-on fee is charged for its processing (S-204), in addition to the S-202 fee. Please note that SCGC does not process biosafety levels higher than 2.
SAG Generation 3 (S-202)
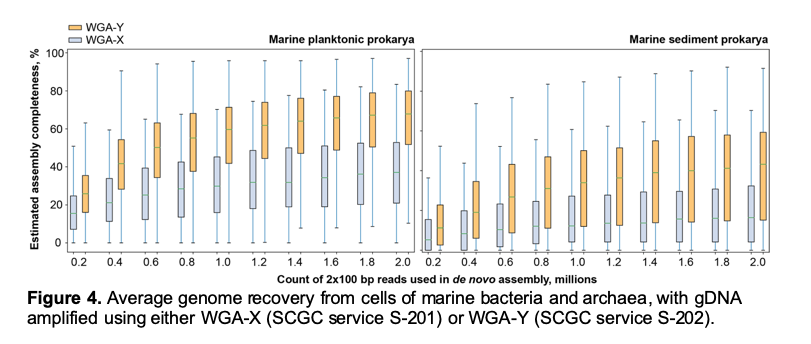
Service S-202 results in the same set of deliverables and involves the same requirements as S-201. The only difference of S-202 from S-201 is a modified genomic DNA amplification technique, WGA-Y, which enables a substantially improved average genome recovery from single cells (Figure 4). Although WGA-Y per se is more expensive than WGA-X, it allows for shallower and cheaper sequencing of the resulting SAGs to produce similar or better genome recovery (Figure 4), which may reduce the overall project costs (see Table 2 below). A manuscript describing the details of WGA-Y is in preparation.
Integrated single cell respiration measurement (S-203)
Service S-203 complements SAG generation (either S-201 or S-202) with the measurement of oxygen respiration rates of individual microbial cells (see reference 13). In order for SCGC to perform this service, customers must provide samples that have been pre-labeled with the RedoxSensor Green probe (Thermo Fisher Scientific), following protocol linked here. Deliverables of S-203 include estimates of O2 respiration rates of the same, individual cells that were used in SAG generation. Please note that this method has not been fully validated for anaerobic respiratory processes.
Biosafety level 2 handling (S-204)
Service S-204 complements services S-201, S-202 and S-203 when processing biosafety level 2 samples. This fee covers the costs of laboratory preparation and staff training for processing biohazardous materials.
SCGC SERVICES: SAG SEQUENCING AND BIOINFORMATICS
SCGC’s SAG whole genome sequencing (WGS) services combine library preparation, shotgun sequencing, de novo assembly, annotation and quality control. Each of these steps are optimized for single cell genomics and validated using benchmark cultures (2). Please note that SCGC does not process SAGs that have been handled elsewhere, in order to prevent the risk of DNA contamination at our facility.
Sequencing libraries are prepared with Nextera XT (Illumina) reagents using a modified protocol (2). Multiplexed libraries are sequenced with Illumina’s NextSeq 2000. SCGC’s de novo genome assembly workflow involves quality-trimming of raw reads with Trimmomatic (5), removal of low-complexity and reagent contaminant reads, read normalization with kmernorm, assembly with SPAdes (6), quality-trimming of contigs, and the removal of contigs shorter than 2000 bp, as previously described (2). Functional annotation is performed using Prokka (7), complemented with a custom protein annotation database built from compiling Swiss-Prot (8) entries for Archaea and Bacteria. SAG taxonomic assignments are based on GTDB-Tk (14) and Silva (15). Estimates of genome assembly completion and potential contamination are obtained with checkM (9). A complementary search for contaminant sequences is performed using tetramer principal component analysis (10). Please note that our current annotation tools are designed for bacterial and archaeal genomes and are not suitable for eukaryotes. Deliverables of all SCGC WGS services include:
a) raw sequence reads
b) de novo SAG assemblies
c) functional annotation of bacterial and archaeal SAGs
d) SAG taxonomic assignments
e) general genome properties, such as GC content and coding density
f) files to assist manual QC: outputs of tetramer PCA, BLASTn and checkM
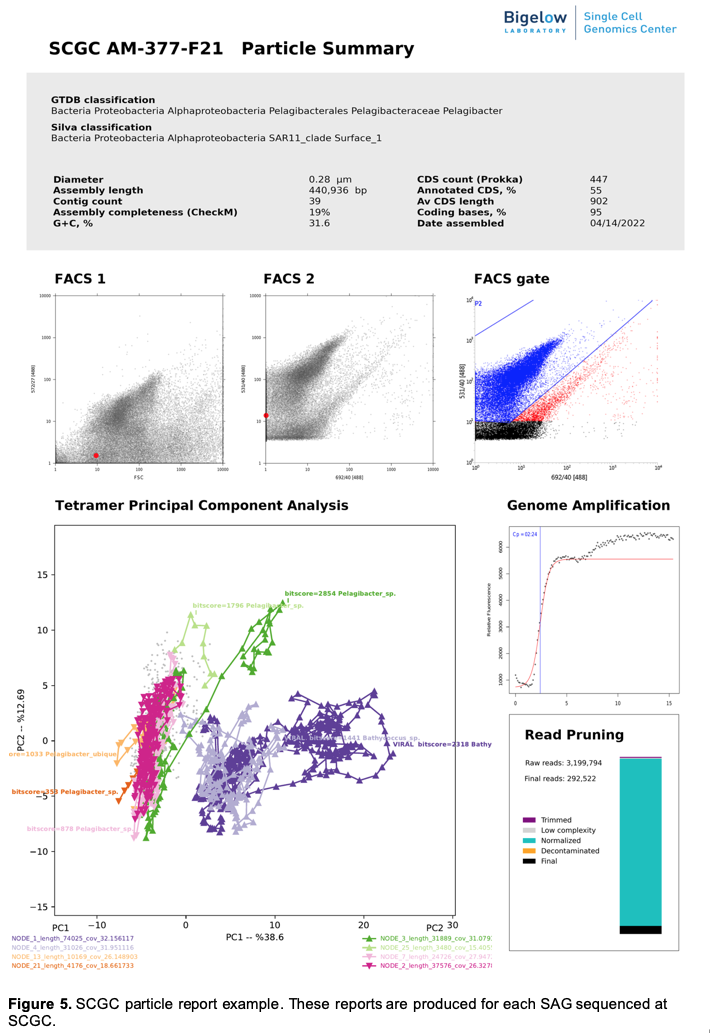
g) Single Particle Reports. These reports bring to your fingertips all key pieces of information about individual cells and other particles analyzed by SCGC, see example in Figure 5.
Specialized laboratory and computational procedures are necessary in order to obtain high quality genomic sequences from SAGs. A particular challenge to consider is the cross-talk of multiplexed libraries (also known as “sample bleeding” and “index switching”) (11, 12). De novo assemblies of SAGs are particularly susceptible to this problem, due to the fact that single cell whole genome amplification is highly uneven across the genome, and deep sequencing is typically employed to facilitate the recovery of the under-amplified genome regions (1). As a result, even a relatively small overall fraction of miss-assigned reads may form contigs that represent over-amplified regions of co-sequenced SAGs. SCGC uses in-house infrastructure and procedures for SAG sequencing that eliminate library cross-talk and do not require subsequent, computational decontamination (2). To verify the efficacy of these solutions, SCGC benchmarks its entire workflow using SAGs of previously sequenced strains (2). We encourage SCGC customers to take similar precautions and workflow validation when sequencing SAGs outside SCGC, in order to maintain data integrity.
SAG WGS 1.8 billion reads (S-211)
This service is best suited for sequencing of large batches of SAGs for maximal genome recovery. DNA sequencing is performed using an Illumina NextSeq2000 instrument and P4 reagents in 2×100 bp mode, resulting in a total of ~1.8 billion paired-end reads. Raw reads are processed using our standard bioinformatics assembly, annotation and QC workflow, as described above. A total of up to 384 SAGs can be selected for sequencing from up to six, 384-well SAG plates generated using SCGC services S-201 or S-202. SAG selection can be done by the customer or by SCGC, e.g.; at random or based on the WGA-X Cp values, which correlate with SAG’s potential for high genome recovery (Figure 2). The average SAG sequencing depth depends on the number of SAGs selected by a customer for sequencing, which can be guided by our prior results with benchmark cultures (Figure 3). Some SAGs may generate low read numbers, due to physical, biological or biochemical factors of individual cells or samples, or due to liquid handling inconsistencies. Due to the high-throughput, low-cost nature of SCGC WGS services, they do not include free re-sequencing of such SAGs, unless the number of affected SAGs exceeds 10% of all sequenced SAGs.
SAG WGS 0.4 billion reads (S-212)
This service is similar to S-211; the only differences are: a) use of P2 sequencing reagents; b) lower cost; and c) the total read count is ~0.4 billion, i.e.; ~3x lower than with service S-211. Service S-212 is best suited for full-depth sequencing of up to 384 prokaryote SAGs produced by S-202 or up to 100 prokaryote SAGs produced by S-201.
SAG WGS 0.1 billion reads (S-216)
This service is similar to S-211; the only differences are: a) use of P1 sequencing reagents; b) lower cost; and c) the total read count is ~0.1 billion, i.e.; ~11x lower than with service S-211. Service S-216 is best suited for full-depth sequencing of small numbers of SAGs or low-coverage sequencing of up to 384 SAGs.
SAG WGS addon 1.8 billion reads (S-213)
This service includes deeper sequencing, de novo assembly, functional annotation and QC of up to 384 SAGs for which compatible Illumina libraries have already been produced by a single service S-211, S-212 or S-216. The total number of new, 2×100 bp reads produced by this service is ~1.8 billion and is distributed relatively evenly among the selected SAGs. More reads per SAG usually results in better genome recovery, but this relationship is not linear (Figure 3). Per customer’s request, SCGC can generate lists of SAGs that meet S-213 eligibility criteria and are either randomized or prioritize SAGs with lowest WGA Cp values – indicators of the potential for high genome recovery (Figure 2). Please note that, due to potential liquid handling inconsistencies, some SAGs may receive unexpectedly low numbers of sequence reads. Due to its high-throughput, low-cost nature, service S-213 does not include free re-sequencing of such SAGs, unless the number of affected SAGs exceeds 10% of all sequenced SAGs.
SAG WGS addon 0.4 billion reads (S-214)
This service is similar to S-213. The only differences are: a) use of P2 sequencing reagents; b) lower cost; and c) the total read count is ~0.4 billion, i.e.; ~3x lower than with service S-213.
SAG WGS addon 0.1 billion reads (S-215)
This service is similar to S-213. The only differences are: a) use of P1 sequencing reagents; b) lower cost; and c) the total read count is ~0.1 billion, i.e.; ~11x lower than with service S-213.
SCGC SERVICES: SEMI-PERMEABLE CAPSULE (SPC) LIBRARY PROCESSING NEW!
SCGC partners with Atrandi Biosciences (Vilnius, Lithuania) in the development of novel, SPC-based technologies to analyze individual cells and other genetic elements. Currently, we offer bioinformatics services that use customer-provided raw reads to produce de novo assembly and annotation of prokarya genomes (service S-301). We also offer sequencing of customer-generated libraries, followed by de novo assembly and prokarya-optimized genome annotation. Only libraries produced using a combination of kits CKP-BARK1 (Atrandi) and E7805S (NEB), following Atrandi instructions, are suitable for these services.
IMPORTANT: Due to the fact that SCGC has no control over the generation of sequencing libraries for these services, SCGC’s quality guarantee is limited to the work performed at SCGC. SCGC’s general Terms & Conditions apply.
SPC library de novo assembly and annotation (S-301)
This service includes computational processing of customer-provided Illumina reads of libraries that were produced using a combination of kits CKP-BARK1 (Atrandi) and E7805S (NEB), following Atrandi instructions. Either raw data from a single Illumina run (BCL files) or demultiplexed reads are acceptable. Service fees are calculated based on dataset size, rounded up to the closest billion PE reads.
SCGC’s computational workflow involves parsing of BCL files into demultiplexed read libraries using Atrandi software (optional); quality-trimming of raw reads with Trimmomatic (5); removal of low-complexity reads with SCGC’s in-house code; read normalization with kmernorm (DOE JGI); de novo assembly with SPAdes (6); prokarya-optimized genome annotation using Prokka (7) and Swiss-Prot (8) database; taxonomic assignments based on GTDB-Tk (14) and Silva (15); and estimates of genome assembly completeness using checkM (9). Several additional analyses are performed in order to facilitate follow-up, manual search for contaminant contigs, including principal component analysis on nucleotide tetramer frequencies (10) and BLASTn against GenBak nr. SCGC’s de novo genome assembly and annotation method description has been published previously (1). This pipeline has been benchmarked for assembly errors using microbial cultures with diverse genome complexity and GC content, indicating no non-target or undefined bases and average frequencies of mis-assemblies, indels and mismatches at <5 per 100 kbp (1). Service deliverables include:
a) demultiplexed sequence reads
b) SPC de novogenome assemblies
c) prokarya-centric functional assembly annotation
d) assembly taxonomic assignments
e) general genome properties, such as GC content and coding density
f) files to assist manual QC: outputs of tetramer PCA, BLASTn and checkM
SPC sequencing, de novo assembly and annotation, 108 reads (S-302)
This service includes sequencing and computational processing of customer-provided libraries that were produced using a combination of kits CKP-BARK1 (Atrandi) and E7805S (NEB), following Atrandi instructions. Minimal library quantity is 4 nM in 30 uL. Customers must provide NEB index primer sequences that were used to generate libraries. Included in this service is one sequencing run using a NextSeq 2000 P1 300-cycle sequencing kit, which typically produces about 0.1 billion paired-end reads. The computational workflow is identical to service S-301, see above.
SPC sequencing, de novo assembly and annotation, 4×108 reads (S-303)
This service is identical to S-302, except that sequencing is done using a NextSeq 2000 P2 300-cycle sequencing kit, which typically produces about 0.4 billion paired-end reads.
SPC sequencing, de novo assembly and annotation, 1.8×109 reads (S-304)
This service is identical to S-302, except that sequencing is done using a NextSeq 2000 P4 300-cycle sequencing kit, which typically produces about 1.8 billion paired-end reads.
SCGC SERVICES: ADDITONAL SERVICES
SAG re-arraying (S-105)
SAGs are transferred from the original 384-well plates to 96-well plates. The SCGC customer defines transfer volumes, source wells, and destination wells. Prior to the transfer, the destination wells are pre-filled with 5-150 uL of either deionized water or 1x TE buffer, as specified by the SCGC customer. Deliverables include re-arrayed SAGs in a 96-well plate.
Sample cryoprotectant glyTE (S-019)
For SCGC customers’ convenience, we offer the preparation and shipment of SCGC’s recommended sample cryoprotectant glyTE. Deliverables of this service include preparation and shipment of 10 mL of 10x concentrated glyTE. Please note that SCGC’s recommended procedures for sample cryopreservation, including the recipe for the cryoprotectant glyTE, are posted on the SCGC website under Preparation and Shipment.
Consultation (S-011)
Basic support is included in the pricing of all SCGC services. When SCGC customers require more extensive help with study design and/or data interpretation (greater than two hours per project), we may request compensation for the associated labor costs via collaborative research grants or consultant fees. We may also request co-authorship on resulting publications for those SCGC scientists who are providing substantial, project-specific intellectual input.
Customized services (S-100)
SCGC offers a wide range of customized services, e.g., non-standard cell sorting, non-SAG sequencing, bioinformatics support, method development, etc. Co-authorship on resulting publications may be requested for those SCGC scientists who are providing substantial, project-specific intellectual input. For more information, please contact SCGC manager Brian Thompson.
COST ESTIMATE: CASE STUDY
Let’s assume an external SCGC customer wants to: a) generate two 384-well plates of prokaryote single amplified genomes (SAGs) using an enhanced gDNA amplification process WGA-Y; b) measure respiration rates of the sorted cells; and c) perform whole genome sequencing of 192 best-amplified SAGs from each of the two plates, as defined by their lowest WGA-Y Cp (Figure 2). Fees for this work are summarized in Table 2. Deliverables include:
- Two 384-well microplates containing SAGs with associated indexFACS and gDNA amplification kinetics data.
- Respiration rate estimates for the sorted cells.
- 384 annotated and QC-ed single cell genome assemblies.
Table 2. Cost estimate for a hypothetical SCGC project.
| Service | Cat. # | Price per unit | # of units | Amount |
| SAG Generation 3 | S-202 | $7,400 | 2 | $14,800 |
| Integrated single cell respiration measurement | S-203 | $900 | 2 | $1,800 |
| SAG WGS 0.4 billion reads | S-212 | $19,000 | 1 | $19,000 |
| Total | $35,600 |
REFERENCES
1. Stepanauskas R. 2012. Single cell genomics: An individual look at microbes. Current Opinion in Microbiology 15:613-620.
2. Stepanauskas R, Fergusson EA, Brown J, Poulton NJ, Tupper B, Labonté JM, Becraft ED, Brown JM, Pachiadaki MG, Povilaitis T, Thompson BP, Mascena CJ, Bellows WK, Lubys A. 2017. Improved genome recovery and integrated cell-size analyses of individual uncultured microbial cells and viral particles. Nat Commun 8:84.
3. Stepanauskas R, Sieracki ME. 2007. Matching phylogeny and metabolism in the uncultured marine bacteria, one cell at a time. Proceedings of the National Academy of Sciences of the United States of America 104:9052-9057.
4. Dean FB, Hosono S, Fang LH, Wu XH, Faruqi AF, Bray-Ward P, Sun ZY, Zong QL, Du YF, Du J, Driscoll M, Song WM, Kingsmore SF, Egholm M, Lasken RS. 2002. Comprehensive human genome amplification using multiple displacement amplification. Proceedings Of The National Academy Of Sciences Of The United States Of America 99:5261-5266.
5. Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30:2114-2120.
6. Nurk S, Bankevich A, Antipov D, et al. 2013. Assembling single-cell genomes and mini-metagenomes from chimeric MDA products. Journal of Computational Biology 20:714-737.
7. Seemann T. 2014. Prokka: rapid prokaryotic genome annotation. Bioinformatics 30:2068-2069.
8. Bateman A, Martin MJ, O’Donovan C, et al. 2017. UniProt: The universal protein knowledgebase. Nucleic Acids Research 45:D158-D169.
9. Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. 2015. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. PeerJ PrePrints 2:e1346.
10. Woyke T, Xie G, Copeland A, González JM, Han C, Kiss H, Saw JH, Senin P, Yang C, Chatterji S, Cheng JF, Eisen JA, Sieracki ME, Stepanauskas R. 2009. Assembling the marine metagenome, one cell at a time. PLoS ONE 4.
11. Mitra A, Skrzypczak M, Ginalski K, Rowicka M. 2015. Strategies for achieving high sequencing accuracy for low diversity samples and avoiding sample bleeding using Illumina platform. PLoS ONE 10.
12. Sinha R, Stanley G, Gulati GS, Ezran C, Travaglini KJ, Wei E, Chan CKF, Nabhan AN, Su T, Morganti RM, Conley SD, Chaib H, Red-Horse K, Longaker MT, Snyder MP, Krasnow MA, Weissman IL. 2017. Index switching causes “spreading-of-signal” among multiplexed samples in Illumina HiSeq 4000 DNA sequencing. bioRxiv:125724.
13. Munson-McGee JH, Lindsay MR, Sintes E, Brown JM, D’Angelo T, Brown J, Lubelczyk LC, Tomko P, Emerson D, Orcutt BN, Poulton NJ, Herndl GJ, Stepanauskas R (2022). Decoupling of respiration rates and abundance in marine prokaryoplankton. Nature 612: 764–770.
14. Parks DH, Chuvochina M, Chaumeil PA, Rinke C, Mussig AJ, Hugenholtz P (2020) A complete domain-to-species taxonomy for Bacteria and Archaea. Nat Biotechnol 38:1079-1086.
15. Quast C, Pruesse E, Gerken J, Peplies J, Yarza P, Yilmaz P, Schweer T, Glöckner FO (2012) The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Research 41:D590-D596.